LINFOMA DE CÉLULAS T ANGIOINMUNOBLÁSTICO CUTANEO SIMULANDO UN PROCESO INFLAMATORIO NO TUMORAL.
Dra. Melisa Ortega *; Dr. Romel Ortega Herrera**; Dr. Pablo Ortega Espinosa***
INTRODUCCIÓN:
Los linfomas periféricos de células T (PTCL) representan un grupo heterogéneo de linfomas no Hodgkin con un fenotipo de células T maduras y características clínicas, patológicas e inmunofenotípicas variables. El linfoma angioinmunoblástico de células T (AITL) es uno de los subtipos más comunes de linfomas de células T periférico, se caracteriza por linfadenopatía generalizada, con frecuentes síntomas B, hepatoesplenomegalia, y mal pronóstico (1).
Los AITL generalmente se presenta como una enfermedad sistémica. Además del compromiso de los ganglios, a menudo involucra sitios extraganglionares, como el bazo, médula ósea, pulmones y piel. La afectación cutánea se presenta aproximadamente en la mitad de los pacientes con AITL, más comúnmente como erupción maculopapular (típicamente, morbiliforme inespecífico o exantematoso). Otras manifestaciones incluyen erupciones de urticaria o purpúrica, lesiones papulovesiculares, lesiones nodulares, placas eritemato-escamosas y lesiones ulceradas. Debido a las manifestaciones dermatológicas tan variadas, la biopsia cutánea debe realizarse en estos pacientes para diferenciar la verdadera afectación por AITL de las condiciones inflamatorias (2)
Presentamos un caso de una paciente con diagnóstico de AITL con lesiones cutáneas, que en otra institución fueron reportadas inicialmente como un proceso inflamatorio no tumoral y luego de ser referida a nuestro laboratorio se pudo determinar infiltración cutánea de AITL.
Además hacemos énfasis en las características histopatológicas del AITL en la biopsia nodal y cutánea, estudios de inmunohistoquímica y correlación clínico patológica.
PRESENTACIÓN DE CASO:
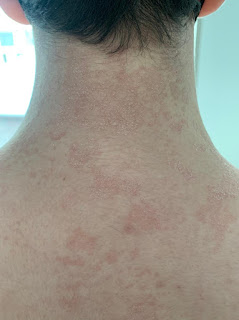
Paciente femenina de 49 años de edad, residente en cantón de la parte alta de la provincia del Oro, presenta linfadenopatías cervicales e inguinales, fijas no dolorosas, que se acompañaban de diaforesis nocturna y malestar general de aproximadamente 2 meses de evolución. Concomitantemente a esta sintomatología la paciente debuta con lesiones cutáneas papulares y nodulares eritematosas localizadas en extremidades superiores, abdomen y tórax anterior, posterior y región glútea. imagen 1 La paciente fue atendida en otra casa de salud en donde mediante análisis de ganglio linfático inguinal fragmentado determinó linfoma nodal de tipo T. Sin embargo, las lesiones cutáneas fueron reportadas como negativas para proceso neoplásico linfomatoso. Al mismo tiempo la paciente acudió otro facultativo quién la diagnosticó de artritis reumatoide para lo cual inició tratamiento a base de corticoides luego de lo cual desapareció la sintomatología ganglionar, malestar general, fiebre nocturna, llegándose a dudar del diagnóstico inicial por el sorprendente buen estado general, y persistencia de lesiones cutáneas que daban la apariencia de ser un proceso inflamatorio no tumoral o infeccioso.
Por este motivo la paciente fue referida a nuestro laboratorio en donde se decidió reevaluar el caso mediante nueva biopsia de piel y de ganglio linfático cervical luego de suspender previamente la administración de corticoides.
Se solicitó a cirugía oncológica realizar biopsia excisional de ganglios linfáticos cervicales recibiéndose en nuestro laboratorio tejido ganglionar íntegro, lo cual nos permitió evaluar su arquitectura histológica a poco aumento, que mostró células atípicas que rebasaba la cápsula del ganglio infiltrando el tejido graso perinodal, con preservación de los sinusoides corticales. Las células neoplásicas fueron de pequeño y mediano tamaño, con citoplasma claro o anfófilo amplio con membranas celulares bien definidas. Las células neoplásicas formaban sábanas o agregados alrededor de las vénulas de endotelios altos lo cuales se mostraron prominentes y numerosos, arborizantes principalmente en la paracorteza. Las células tumorales estaban acompañadas de una población inflamatoria polimorfa compuesta por linfocitos reactivos, histiocitos, células plasmáticas y frecuentes eosinófilos.
La biopsia de piel fue realizada en nuestro laboratorio y mostró epidermis de aspecto histológico conservado sin epidermotropismo. Con respecto a la dermis superficial y profunda se observó denso infiltrado perivascular y perianexial prominentes compuestos por células atípicas de pequeño y mediano tamaño, de núcleos con pleomorfismo moderado, cromatina fina y nucleolos discretos, y de citoplasma amplio claro bien definido distribuido principalmente alrededor de los casos capilares y glándulas ecrinas. imagen 2.
Las células tumorales estaba entremezcladas con una población inflamatoria mixta compuesta por linfocitos, histiocitos, células plasmáticas y numerosos eosinófilos. imagen 3 Las tinciones de inmunohistoquímica mostraron positividad para el CD4, CD10, BCL-6, Epstein Barr virus en forma focal. imagen 4. Los hallazgos histológicos y de inmunohistoquímica fueron concluyentes con "linfoma de células T angioinmunoblástico nodal con extensión a piel".







DISCUSIÓN:
En cuanto a la inmunohistoquímica, el AITL expresa antígenos de células Pan T como el CD2, CD3, CD5 y CD7 con pérdida aberrante frecuente de uno o más de estos antígenos. El marcador de células T colaboradoras CD4 se expresa de forma más consistente. También existe expresión de marcadores foliculares de células T colaboradoras con son el CD10, BCL6, CXCL13 (más específico), PD-1 (CD279), marcadores de células dendríticas foliculares como el CD21, CD23, CD35, o clusterina y algunas células B dispersas expresan EBV +.
Recientemente se ha descrito que la afectación a piel por AITL presenta características histopatológicas no muy claras, y que simula en la mayoría de los casos un proceso inflamatorio no tumoral (6) o infeccioso, y cuando existe numerosos histiocitos epitelioides puede incluso simular una enfermedad de Hansen (9). En el presente caso se realizaron tinciones de PAS y Ziehl Neelsen que fueron negativas para hongos y bacilos ácido alcohol resistentes respectivamente, descartando con esto un proceso infeccioso.
Histopatológicamente, la afectación cutánea por AITL se caracteriza por una infiltración perivascular de células T de tamaño pequeño a mediano, con epidermotropismo mínimo o nulo, así como histiocitos reactivos, células plasmáticas y/o eosinófilos. Sin embargo, los hallazgos característicos de AITL en ganglio como linfocitos atípicos con citoplasma amplio y capilares hiperplásicos están presentes solo en la minoría de biopsias cutáneas, enfatizando que los criterios de diagnóstico para AITL ganglionar pueden no ser totalmente aplicable a sitios cutáneos. En particular, las lesiones que consisten en una infiltración linfocítica perivascular escasa sin atipia obvia pueden mostrar características inespecíficas y ser difíciles de diferenciar de enfermedades inflamatorias o infecciosas (6).
Cabe mencionar que el diagnóstico de linfomas cutáneos de células T es complejo, y requiere tener experiencia, además de sólidos conocimientos dermatopatológicos, inmunohistoquímicos y clínicos, para interpretar y diagnosticar este tipo de linfomas. Por lo tanto, es conveniente realizar una adecuada correlación clínico patológica y el análisis del ganglio linfático en estos pacientes, tomando en cuenta que algunas pruebas de laboratorio pueden confundir con enfermedades inmunológicas, como fue el caso de nuestra paciente.
Además, recomendamos realizar una segunda toma de biopsia de piel y ganglio linfático en casos en los cuales el diagnóstico es sombrío o dudoso, siendo una práctica apropiada y que brinda información valiosa, como fue el caso de nuestra paciente en la cual se llegó a determinar infiltración nodal y cutánea por AITL en un segundo análisis de biopsia.
Finalmente, la importancia de establecer infiltración neoplásica cutánea en pacientes con AITL nodal radica en que ensombrece el pronóstico de sobrevida, requiriéndose esquemas de tratamiento diferentes a los que se utilizan en estadíos más tempranos de la enfermedad (10).

Comentarios
Publicar un comentario